


Sickle Cell Disease Explained: A Guide to Genetic Blood Disorders
Sickle cell disease (SCD) is a genetic blood disorder that affects approximately 100,000 Americans, making it the most common genetic blood disorder in the country.
Understanding Sickle Cell Disease
Sickle cell disease is a hereditary condition caused by a mutation in the genes that encode hemoglobin, the oxygen-carrying protein in red blood cells. This mutation leads to the production of an abnormal form of hemoglobin called hemoglobin S (HgbS).
Genetics of Sickle Cell Disease
SCD is inherited in an autosomal recessive pattern, meaning a child needs to inherit two copies of the sickle cell gene—one from each parent—to have the disease. Carriers, who inherit only one copy, typically do not show symptoms but can pass the gene to their children.
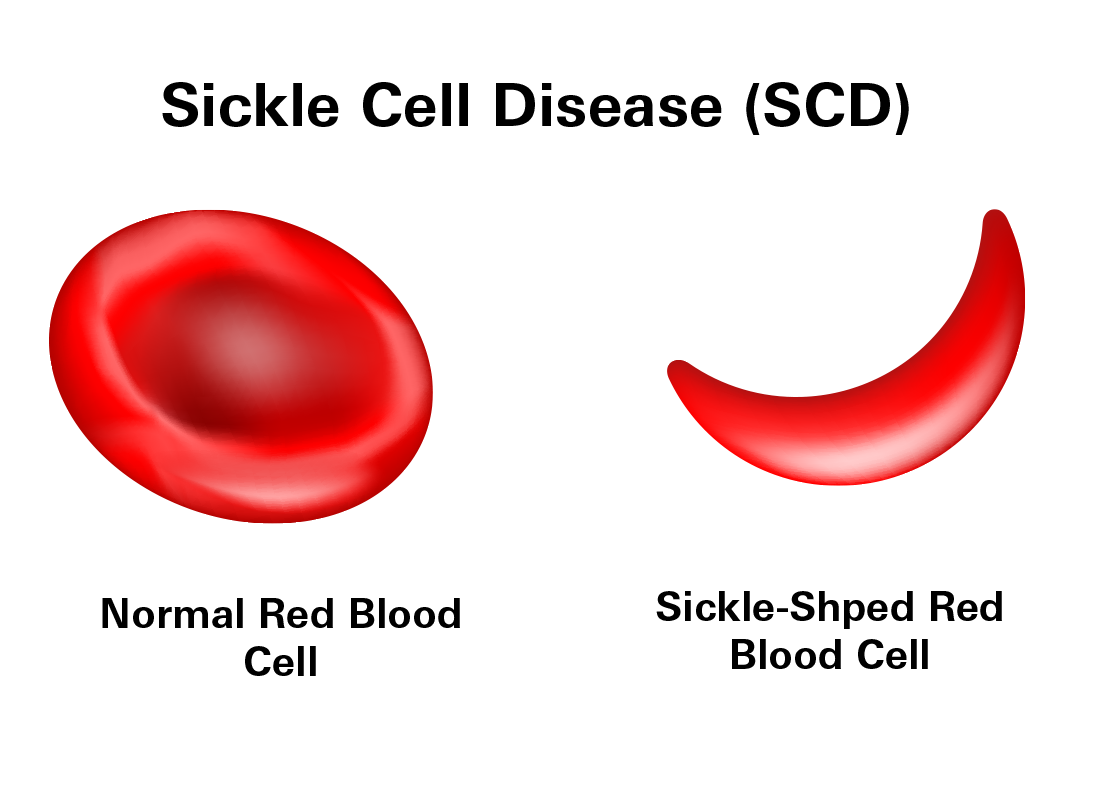
In healthy individuals, red blood cells are round and flexible, which allows them to travel through small blood vessels to deliver oxygen to all parts of the body. However, in SCD, the red blood cells become rigid and sticky and take on a crescent or "sickle" shape due to the abnormal hemoglobin.
These sickle-shaped red blood cells are prone to breaking apart (hemolyzing) after only 10 to 20 days, compared to the normal 90-120 days. This rapid breakdown of red blood cells leads to anemia and fatigue due to a reduced number of red blood cells. The misshapen cells can also stick together and block blood flow in smaller vessels, leading to pain and potential damage to organs.
Newborn Screening
Most babies in the United States and many other countries are screened for sickle cell disease soon after birth as part of standard newborn screening programs. This early detection is vital because it allows for immediate initiation of treatment and management strategies to prevent complications. The screening involves taking a small blood sample from the baby's heel to test for the presence of abnormal hemoglobin.
Signs and Symptoms of Sickle Cell Disease
Sickle cell disease symptoms typically start to appear when a baby is about 2 to 3 months old. This timing is due to the shift from fetal hemoglobin (HgbF), which is present in newborns and helps prevent red blood cells from sickling, to adult hemoglobin that includes the abnormal hemoglobin S found in those with sickle cell disease.
- Anemia: Characterized by extreme fatigue and shortness of breath because the sickle-shaped cells are less efficient at carrying oxygen.
- Pain Episodes: These occur when sickle-shaped red blood cells block blood flow to joints, abdomen, and chest, leading to severe and sometimes acute pain.
- Jaundice: The skin and eyes may appear yellow due to the breakdown of red blood cells, which produces bilirubin.
- Frequent Infections: The spleen, which helps filter bacteria from the blood, may be damaged by sickle cells. This damage makes individuals more susceptible to infections. Children with sickle cell disease often receive antibiotics and vaccinations to prevent life-threatening illnesses like pneumonia. Common pathogens include Salmonella, S. pneumoniae, H. influenzae, and N. meningitidis, which can cause severe infections like sepsis and meningitis.
- Delayed Growth: Children with sickle cell disease may experience slowed growth and delayed puberty. Healthy red blood cells are crucial for delivering the oxygen and nutrients needed for growth.
- Vision Problems and Headaches: These can occur due to blood vessel blockages affecting the eyes and brain.
Important Precautions
- Vaccinations are crucial to protect against infections.
- Monitor for fever: If your temperature exceeds 101.3 °F, seek medical attention promptly as this could be a sign of a serious infection.
Treatment of Sickle Cell Disease
Sickle cell disease treatment varies but focuses on managing symptoms and preventing complications. Here's an overview of the primary treatments available:
Bone Marrow Transplantation
Bone marrow transplantation is currently the only cure for sickle cell disease. However, it's not commonly used due to significant risks, high costs, and the invasive nature of the procedure. Children are more likely to successfully undergo a transplant than adults, who may have accumulated organ damage.
Blood Transfusions
Blood transfusions supplement the patient's supply of red blood cells, helping to prevent complications such as stroke, acute chest syndrome, and severe anemia. Despite their benefits, one risk of regular transfusions is iron overload (hemosiderosis), which can harm organs.
Immunization
Vaccinations are critical, especially for children with impaired spleen function, which increases infection risks. Recommended vaccines include:
- Haemophilus influenzae type b (Hib) and PCV13 (Prevnar) for all children.
- PPSV23 (Pneumovax 23) for children over 2 years.
- Bexsero and Trumenba for children over 10 years.
Medications
- Antibiotics: To prevent infections, daily penicillin (twice daily) is advised for children with sickle cell disease from birth until the age of 5.
- Pain Management: Various analgesics are used to manage pain episodes associated with the disease.
- Hydroxyurea (Droxia, Hydrea): This medication is used to prevent and reduce the frequency of complications caused by sickle cell disease. Hydroxyurea is a disease-modifying agent that stimulates the production of HgbF. Long-term use of Hydroxyurea decreases the frequency of acute pain crises, ep-isodes of acute chest pain syndrome, and the need for blood transfusions.
- L-glutamine (Endari): Approved for use in adults and children over 5, this medication helps to reduce the frequency of sickle cell complications.
These treatments help manage the disease and improve the quality of life for those affected by sickle cell disease. Regular medical supervision and personalized treatment plans are crucial for managing the disease effectively.